Page Not Found
Page not found. Your pixels are in another canvas.

A list of all the posts and pages found on the site. For you robots out there is an XML version available for digesting as well.
Page not found. Your pixels are in another canvas.
About me
This is a page not in th emain menu
Published:
This post will show up by default. To disable scheduling of future posts, edit config.yml and set future: false.
Published:
This is a sample blog post. Lorem ipsum I can’t remember the rest of lorem ipsum and don’t have an internet connection right now. Testing testing testing this blog post. Blog posts are cool.
Published:
This is a sample blog post. Lorem ipsum I can’t remember the rest of lorem ipsum and don’t have an internet connection right now. Testing testing testing this blog post. Blog posts are cool.
Published:
This is a sample blog post. Lorem ipsum I can’t remember the rest of lorem ipsum and don’t have an internet connection right now. Testing testing testing this blog post. Blog posts are cool.
Published:
This is a sample blog post. Lorem ipsum I can’t remember the rest of lorem ipsum and don’t have an internet connection right now. Testing testing testing this blog post. Blog posts are cool.
Since the outbreak of COVID-19 pandemic in 2019, I’m leading a large-scale project, on a national scale, to comprehensively collect multi-omics dataset, including Whole-genome sequencing(WGS), HLA typing, bulk T/BCR-seq, single cell RNA sequencing (scRNA-seq) with T/BCR-seq at a single cell resolution, and expression profiling for 192 cytokines, as well as clinical data and laboratory testing data, through collaboration with 7 distinct hospitals in South Korea. Biospecimen, such as Peripheral blood mononuclear cells (PBMC), Plasma, Serum, Urine and NPS, is also being collected and deposited at National Biobank in Korea, and is distributed for approved researchers. Currently, additional proteomic and epigenetic data is being actively collected to make the immunological mechanisms by SARS-CoV-2 understand in detail, which , in turn, prevent upcoming virus in the future. It is expected that this project would provide invaluable insights and discoveries in the era of crisis caused by infectious diseases and understand detailed immunology in terms of host defensive mechanisms throughput multi-layer data. 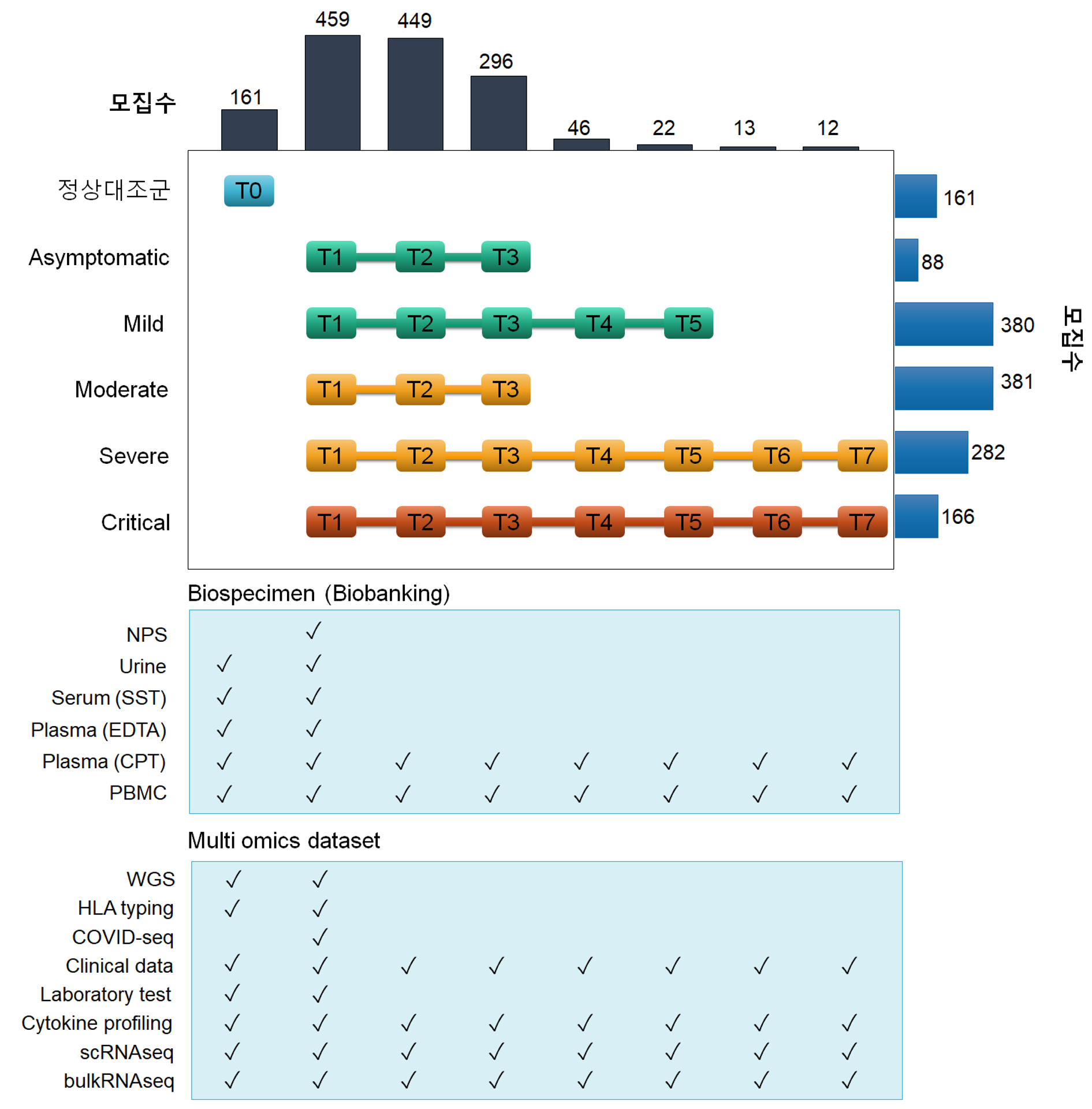
Severity prediction against COVID-19 is crucial to perform early screening, detection and stratification of patients with COVID- 19. In this study, I, as a principle investigator (PI), developed machine learning (ML) models that predict COVID-19 severity based on clinical and laboratory testing data from 300 COVID-19 patients and 120 healthy controls. The proposed ML model with the selected features showed accurate prediction of COVID-19 severity. It is expected that this results provide evidence for not only efficient data- driven decision support to clinicians in urgent COVID-19 diagnostic situations, but also early detection and stratification of COVID-19 severity. Further, this study would be expanded into integrative approaches based on heterogeneous dataset, including high-dimensional omics to figure out remarkable markers for COVID-19 severity. 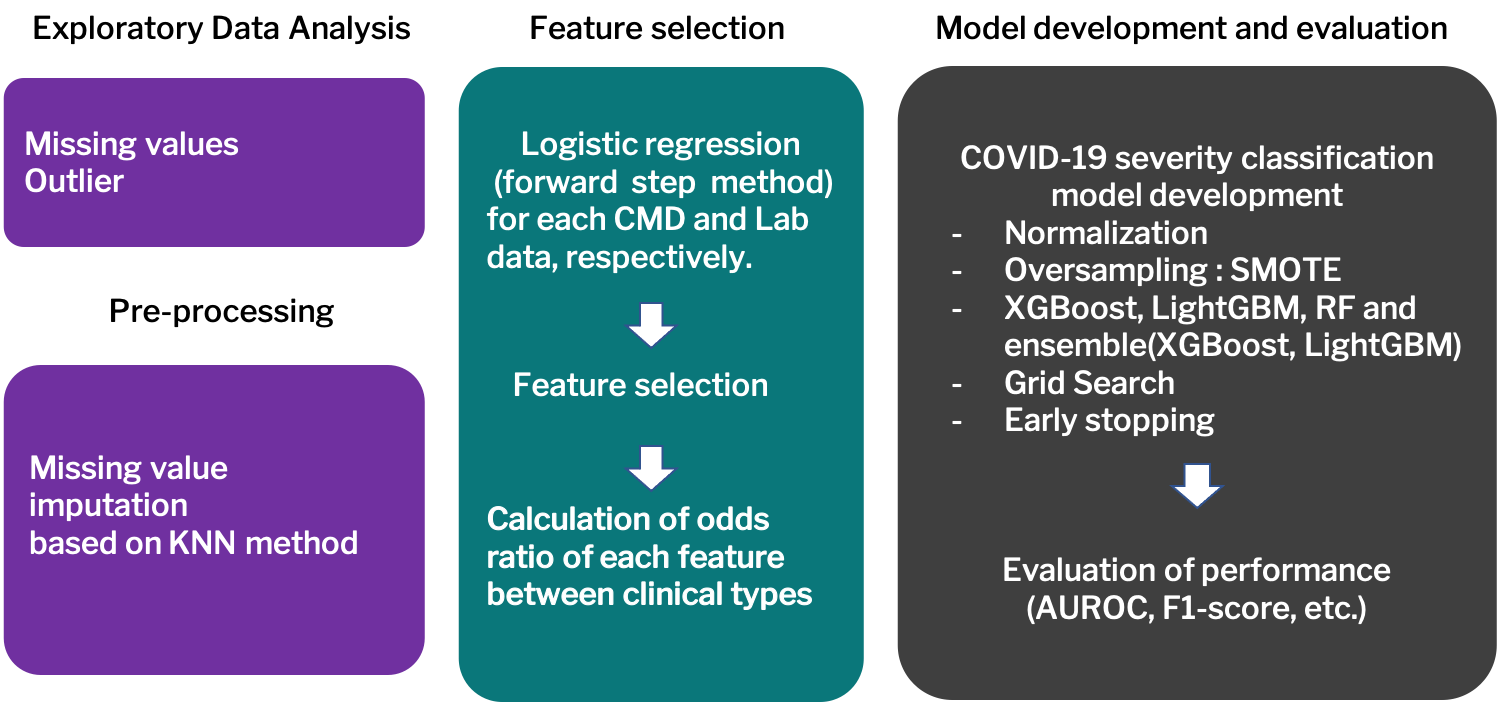
Published in Clinical Genetics, 2016
Large insertions and deletions (indels), including copy number variations (CNVs), are commonly seen in many diseases. Standard approaches for indel detection rely on well-established methods such as qPCR or short tandem repeat (STR) markers. Recently, a number of tools for CNV detection based on next-generation sequencing (NGS) data have also been developed; however, use of these methods is limited. Here, we used whole-exome sequencing (WES) in patients previously diagnosed with CMT1A or HNPP using STR markers to evaluate the ability of WES to improve the clinical diagnosis. Patients were evaluated utilizing three CNV detection tools including CONIFER, ExomeCNV and CEQer, and array comparative genomic hybridization (aCGH). We identified a breakpoint region at 17p11.2-p12 in patients with CMT1A and HNPP. CNV detection levels were similar in both 6 Gb (mean read depth = 80×) and 17 Gb (mean read depth = 190×) data. Taken together, these data suggest that 6 Gb WES data are sufficient to reveal the genetic causes of various diseases and can be used to estimate single mutations, indels, and CNVs simultaneously. Furthermore, our data strongly indicate that CNV detection by NGS is a rapid and cost-effective method for clinical diagnosis of genetically heterogeneous disorders such as CMT neuropathy.
Recommended citation: Hye-Yeong Jo et al.,. (2016). "Application of whole-exome sequencing for detecting copy number variants in CMT1A/HNPP." Journal 1. 1(2). http://hyeyeongJo.github.io/files/publication1.pdf
Published in PLoS One, 2017
Insertion and deletion (INDEL) mutations, the most common type of structural variance, are associated with several human diseases. The detection of INDELs through next-generation sequencing (NGS) is becoming more common due to the decrease in costs, the increase in efficiency, and sensitivity improvements demonstrated by the various sequencing platforms and analytical tools. However, there are still many errors associated with INDEL variant calling, and distinguishing INDELs from errors in NGS remains challenging. To evaluate INDEL calling from whole-exome sequencing (WES) data, we performed Sanger sequencing for all INDELs called from the several calling algorithm. We compared the performance of the four algorithms (i.e. GATK, SAMtools, Dindel, and Freebayes) for INDEL detection from the same sample. We examined the sensitivity and PPV of GATK (90.2 and 89.5%, respectively), SAMtools (75.3 and 94.4%, respectively), Dindel (90.1 and 88.6%, respectively), and Freebayes (80.1 and 94.4%, respectively). GATK had the highest sensitivity. Furthermore, we identified INDELs with high PPV (4 algorithms intersection: 98.7%, 3 algorithms intersection: 97.6%, and GATK and SAMtools intersection INDELs: 97.6%). We presented two key sources of difficulties in accurate INDEL detection: 1) the presence of repeat, and 2) heterozygous INDELs. Herein we could suggest the accessible algorithms that selectively reduce error rates and thereby facilitate INDEL detection. Our study may also serve as a basis for understanding the accuracy and completeness of INDEL detection.
Recommended citation: Bo-Young Kim et al., (2017). "Optimized detection of insertions/deletions (INDELs) in whole-exome sequencing data." PLoS One1. 12(8). http://hyeyeongJo.github.io/files/publication2.pdf
Published in BMC Medical Genetics, 2017
Background: Multiple endocrine neoplasia type 1 (MEN1) syndrome is an autosomal dominant hereditary disorder characterized by the presence of endocrine tumors affecting the parathyroid, pancreas, and pituitary. A heterozygous germline inactivating mutation in the MEN1 gene (first hit) may be followed by somatic loss of the remaining normal copy or somatic mutations in the MEN1 gene (second hit). Whole-exome sequencing has been successfully used to elucidate the mutations associated with the different types of tumors. Case presentation: We performed whole-exome sequencing (WES) on three parathyroid tumors, one pancreatic insulinoma, and a blood sample taken from the same patient with MEN1 to study tumor heterogeneity in MEN1 originating from different tumors. We identified a novel frame-shift deletion (c.1382_1383delAG, p.E461GfsX69) in the MEN1 gene using WES, which was confirmed by Sanger sequencing. WES and the SNP array revealed somatic LOH on chromosome 11 in parathyroid tumors (left upper, left lower, and right upper parathyroid). However, we did not detect a somatic MEN1 gene mutation or LOH in the pancreatic insulinoma. WES revealed two somatic functional variants outside the MEN1 gene in the pancreatic insulinoma. Conclusions: This study revealed heterogeneity among tumors in the same patient with MEN1, suggesting that different tumor-specific tumorigenic mechanisms may contribute to the pathogenesis of MEN1 tumors. The present study supports the clinical applicability of the WES strategy to research on multiple tumor samples and blood.
Recommended citation: Bo-Young Kim et al., (2017). "Genetic analysis of parathyroid and pancreatic tumors in a patient with multiple endocrine neoplasia type 1 using whole-exome sequencing." BMC Medical Genetics. 18(1):106. http://hyeyeongJo.github.io/files/publication3.pdf
Published in Biotechnology Letters, 2019
A major limitation in anti-tuberculosis drug screening is the lack of reliable and scalable models for homogeneous human primary macrophage cells of non-cancer origin. Here we report a modified protocol for generating homogeneous populations of macrophage-like cells from human embryonic stem cells. The induced macrophages, referred to as iMACs, presented similar transcriptomic profiles and characteristic immunological features of classical macrophages and were permissive to viral and bacterial infection, in particular Mycobacterium tuberculosis (Mtb). More importantly, iMAC production was amenable to scale up. To evaluate iMAC efficiency in high-throughput anti-tuberculosis drug screening, we performed a phenotypic screening against intracellular Mtb, involving a library of 3,716 compounds that included FDA-approved drugs and other bioactive compounds. Our primary screen identified 120 hits, which were validated in a secondary screen by dose-intracellular and -extracellular Mtb assays. Our confirmatory studies identified a novel anti-Mtb compound, 10-DEBC, also showing activity against drug-resistant strains.
Recommended citation: H-Y Jo et al.,. (2017). "Peroxide-dependent oxidation reactions catalyzed by CYP191A1 from Mycobacterium smegmatis." Biotechnology Letters. 1(2). http://hyeyeongJo.github.io/files/publication4.html
Published in Stem Cell Reports, 2019
A major limitation in anti-tuberculosis drug screening is the lack of reliable and scalable models for homogeneous human primary macrophage cells of non-cancer origin. Here we report a modified protocol for generating homogeneous populations of macrophage-like cells from human embryonic stem cells. The induced macrophages, referred to as iMACs, presented similar transcriptomic profiles and characteristic immunological features of classical macrophages and were permissive to viral and bacterial infection, in particular Mycobacterium tuberculosis (Mtb). More importantly, iMAC production was amenable to scale up. To evaluate iMAC efficiency in high-throughput anti-tuberculosis drug screening, we performed a phenotypic screening against intracellular Mtb, involving a library of 3,716 compounds that included FDA-approved drugs and other bioactive compounds. Our primary screen identified 120 hits, which were validated in a secondary screen by dose-intracellular and -extracellular Mtb assays. Our confirmatory studies identified a novel anti-Mtb compound, 10-DEBC, also showing activity against drug-resistant strains.
Recommended citation: Hye-Yeong Jo et al.,. (2019). "Drug Discovery Platform Targeting M. tuberculosis with Human Embryonic Stem Cell-Derived Macrophages." Stem Cell Reports. 13(6):980-991. http://hyeyeongJo.github.io/files/publication5.pdf
Published in Biochem Biophys Res Commun, 2020
Assessment of differentiation potential is a basic requirement to obtain qualified human pluripotent stem cells (hPSCs). Here, we report a simple differentiation method using fetal bovine serum (FBS) to estimate differentiation potential and propensity of hPSCs. PluriTest using RNA-sequencing showed that cells differentiated after treatment with 5% FBS. Expression patterns of three germ layer markers revealed that cells cultured in Knockout Serum Replacement-containing medium (KSR) with mouse feeder cells had higher differentiation potential than cells cultured in a chemically defined medium (E8) with recombinant matrix proteins, especially into the mesoderm and endoderm lineages. Analysis of differentially expressed genes between KSR and E8 identified DUSP6 as a marker for where cells had been cultured. Expression of DUSP6 correlated with FGF-ERK signaling activity. Fine-tuning of FGF-ERK signaling activity to a range that can shut down DUSP6 transcription but sustain NANOG transcription partially increased the differentiation potential. Our data suggest that differentiation with 5% FBS is good to estimate differentiation potential and propensity at the early stage, and that DUSP6 is an excellent marker to monitor ERK signaling activity.
Recommended citation: DH Yoo et al.,. (2020). "Simple differentiation method using FBS identifies DUSP6 as a marker for fine-tuning of FGF-ERK signaling activity in human pluripotent stem cells." Biochem Biophys Res Commun. 521(2):375-382. http://hyeyeongJo.github.io/files/publication6.pdf
Published in Scientific Reports, 2020
Although human induced pluripotent stem cell (hiPSC) lines are karyotypically normal, they retain the potential for mutation in the genome. Accordingly, intensive and relevant quality controls for clinical-grade hiPSCs remain imperative. As a conceptual approach, we performed RNA-seq-based broad-range genetic quality tests on GMP-compliant human leucocyte antigen (HLA)-homozygous hiPSCs and their derivatives under postdistribution conditions to investigate whether sequencing data could provide a basis for future quality control. We found differences in the degree of single-nucleotide polymorphism (SNP) occurring in cells cultured at three collaborating institutes. However, the cells cultured at each centre showed similar trends, in which more SNPs occurred in late-passage hiPSCs than in early-passage hiPSCs after differentiation. In eSNP karyotyping analysis, none of the predicted copy number variations (CNVs) were identified, which confirmed the results of SNP chip-based CNV analysis. HLA genotyping analysis revealed that each cell line was homozygous for HLA-A, HLA-B, and DRB1 and heterozygous for HLA-DPB type. Gene expression profiling showed a similar differentiation ability of early- and late-passage hiPSCs into cardiomyocyte-like, hepatic-like, and neuronal cell types. However, time-course analysis identified five clusters showing different patterns of gene expression, which were mainly related to the immune response. In conclusion, RNA-seq analysis appears to offer an informative genetic quality testing approach for such cell types and allows the early screening of candidate hiPSC seed stocks for clinical use by facilitating safety and potential risk evaluation.
Recommended citation: Hye-Yeong Jo et al.,. (2020). "Development of genetic quality tests for good manufacturing practice-compliant induced pluripotent stem cells and their derivatives." Scientific Reports. 10(1):3939. http://hyeyeongJo.github.io/files/publication7.pdf
Published in Scientific Reports, 2020
Human pluripotent stem cells (hPSCs) have promising therapeutic applications due to their infinite capacity for self-renewal and pluripotency. Genomic stability is imperative for the clinical use of hPSCs; however, copy number variation (CNV), especially recurrent CNV at 20q11.21, may contribute genomic instability of hPSCs. Furthermore, the effects of CNVs in hPSCs at the whole-transcriptome scale are poorly understood. This study aimed to examine the functional in vivo and in vitro effects of frequently detected CNVs at 20q11.21 during early-stage differentiation of hPSCs. Comprehensive transcriptome profiling of abnormal hPSCs revealed that the differential gene expression patterns had a negative effect on differentiation potential. Transcriptional heterogeneity identified by single-cell RNA sequencing (scRNA-seq) of embryoid bodies from two different isogenic lines of hPSCs revealed alterations in differentiated cell distributions compared with that of normal cells. RNA-seq analysis of 22 teratomas identified several differentially expressed lineage-specific markers in hPSCs with CNVs, consistent with the histological results of the altered ecto/meso/endodermal ratio due to CNVs. Our results suggest that CNV amplification contributes to cell proliferation, apoptosis, and cell fate specification. This work shows the functional consequences of recurrent genetic abnormalities and thereby provides evidence to support the development of cell-based applications.
Recommended citation: Hye-Yeong Jo et al.,. (2020). "Functional in vivo and in vitro effects of 20q11.21 genetic aberrations on hPSC differentiation." Stem Cell Reports. 10(1):18582. http://hyeyeongJo.github.io/files/publication8.pdf
Published in Stem Cell Research, 2021
The Korea National Stem Cell Bank has been banking pluripotent stem cell (PSC) lines since 2012. Quality-controlled and ethically sourced cell lines were developed for distribution. Currently (as of 2020), among the 69 deposited lines, 4 research-grade human embryonic stem cell (hESC) lines and 19 induced pluripotent stem cell (iPSC) lines have been distributed. Good manufacturing practices (GMP)-compliant homozygous iPSC lines for regenerative medicine with homozygous HLA haplotypes that cover 51% of the Korean population have been deposited as well. To ensure the quality of the cell lines, we performed eighteen different quality tests on the identity, sterility, consistency, stability and safety of the cell lines. Regarding genetic stability, we are collecting SNPchip, WES, Methyl-seq, and RNA-seq data, which are open to the public.s.
Recommended citation: Jung-Hyun Kim et al.,. (2021). "Korea National Stem Cell Bank." Stem Cell Research. 53:102270. http://hyeyeongJo.github.io/files/publication9.pdf
Published in Frontiers in Genetics, 2022
Macrophages exhibit high plasticity to achieve their roles in maintaining tissue homeostasis, innate immunity, tissue repair and regeneration. Therefore, macrophages are being evaluated for cell-based therapeutics against inflammatory disorders and cancer. To overcome the limitation related to expansion of primary macrophages and cell numbers, human pluripotent stem cell (hPSC)-derived macrophages are considered as an alternative source of primary macrophages for clinical application. However, the quality of hPSC-derived macrophages with respect to the biological homogeneity remains still unclear. We previously reported a technique to produce hPSC-derived macrophages referred to as iMACs, which is amenable for scale-up. In this study, we have evaluated the biological homogeneity of the iMACs using a transcriptome dataset of 6,230 iMACs obtained by single-cell RNA sequencing. The dataset provides a valuable genomic profile for understanding the molecular characteristics of hPSC-derived macrophage cells and provide a measurement of transcriptomic homogeneity. Our study highlights the usefulness of single cell RNA-seq data in quality control of the cell-based therapy products.
Recommended citation: Hye-Yeong Jo et al.,. (2022). "Single-Cell RNA Sequencing of Human Pluripotent Stem Cell-Derived Macrophages for Quality Control of The Cell Therapy Product." Frontiers in Genetics. 12:658862. http://hyeyeongJo.github.io/files/publication10.pdf
Published in BMB Reports, 2022
Understanding and monitoring virus-mediated infections has gained importance since the global outbreak of the coronavirus disease 2019 (COVID-19) pandemic. Studies of high-throughput omics-based immune profiling of COVID-19 patients can help manage the current pandemic and future virus-mediated pandemics. Although COVID-19 is being studied since past 2 years, detailed mechanisms of the initial induction of dynamic immune responses or the molecular mechanisms that characterize disease progression remains unclear. This study involved comprehensively collected biospecimens and longitudinal multi-omics data of 300 COVID-19 patients and 120 healthy controls, including whole genome sequencing (WGS), single-cell RNA sequencing combined with T cell receptor (TCR) and B cell receptor (BCR) sequencing (scRNA(+scTCR/BCR)-seq), bulk BCR and TCR sequencing (bulk TCR/BCR-seq), and cytokine profiling. Clinical data were also collected from hospitalized COVID-19 patients, and HLA typing, laboratory characteristics, and COVID-19 viral genome sequencing were performed during the initial diagnosis. The entire set of biospecimens and multi-omics data generated in this project can be accessed by researchers from the National Biobank of Korea with prior approval. This distribution of largescale multi-omics data of COVID-19 patients can facilitate the understanding of biological crosstalk involved in COVID-19 infection and contribute to the development of potential methodologies for its diagnosis and treatment.
Recommended citation: Hye-Yeong Jo et al., (2022). "Establishment of the large-scale longitudinal multi-omics dataset in COVID-19 patients: data profile and biospecimen." Stem Cell Reports. 55(9):465-471. http://hyeyeongJo.github.io/files/publication11.pdf
Published in Frontiers in Immunology, 2023
Introduction: Despite of massive endeavors to characterize inflammation in COVID-19 patients, the core network of inflammatory mediators responsible for severe pneumonia stillremain remains elusive. Methods: Here, we performed quantitative and kinetic analysis of 191 inflammatory factors in 955 plasma samples from 80 normal controls (sample n = 80) and 347 confirmed COVID-19 pneumonia patients (sample n = 875), including 8 deceased patients. Results: Differential expression analysis showed that 76% of plasmaproteins (145 factors) were upregulated in severe COVID-19 patients comparedwith moderate patients, confirming overt inflammatory responses in severe COVID-19 pneumonia patients. Global correlation analysis of the plasma factorsrevealed two core inflammatory modules, core I and II, comprising mainly myeloid cell and lymphoid cell compartments, respectively, with enhanced impact in a severity-dependent manner. We observed elevated IFNA1 and suppressed IL12p40, presenting a robust inverse correlation in severe patients, which was strongly associated with persistent hyperinflammation in 8.3% of moderate pneumonia patients and 59.4% of severe patients. Discussion: Aberrant persistence of pulmonary and systemic inflammation might be associated with long COVID-19 sequelae. Our comprehensive analysis of inflammatory mediators in plasmarevealed the complexity of pneumonic inflammation in COVID-19 patients anddefined critical modules responsible for severe pneumonic progression.
Recommended citation: Kyeongseok Jeon et al.,. (2023). "Elevated IFNA1 and suppressed IL12p40 associated with persistent hyperinflammation in COVID-19 pneumonia." Frontiers in Immunology. 14:1101808. http://hyeyeongJo.github.io/files/publication12.pdf
Published:
This is a description of your talk, which is a markdown files that can be all markdown-ified like any other post. Yay markdown!
Published:
This is a description of your conference proceedings talk, note the different field in type. You can put anything in this field.
Undergraduate course, University 1, Department, 2014
This is a description of a teaching experience. You can use markdown like any other post.
Workshop, University 1, Department, 2015
This is a description of a teaching experience. You can use markdown like any other post.